

Mutations in the kinase binding domain of BTK at position C481 are associated with resistance to BTK inhibitor (BTKi) therapy in chronic lymphocytic leukemia (CLL). Nearly half of patients manifesting clinical progression with these alterations exhibit a subclonal burden of resistance. Intriguingly, measured BTKC481 variant allelic fractions (VAF) are commonly lower than 10% [Ahn et al, Blood 2017]. This raises the important question of how BTKC481-mutated (MUT) and wildtype (WT) cells differ in their response to therapeutic challenge, and how low-burden MUT subclones facilitate escape from therapy.
While the admixture of MUT and WT cells within the same individual presents an opportunity to directly study the downstream effects of subclonal resistance mutations, these cells cannot be separated through sorting. To overcome this limitation, we utilized Genotyping of Transcriptomes (GoT)-a strategy to jointly capture genotyping of a locus of interest together with whole transcriptomes at the single cell level (Fig. 1A). Importantly, GoT eliminates patient-specific and technical confounders, enabling direct linkage of BTK genotypes to transcriptional phenotypes.
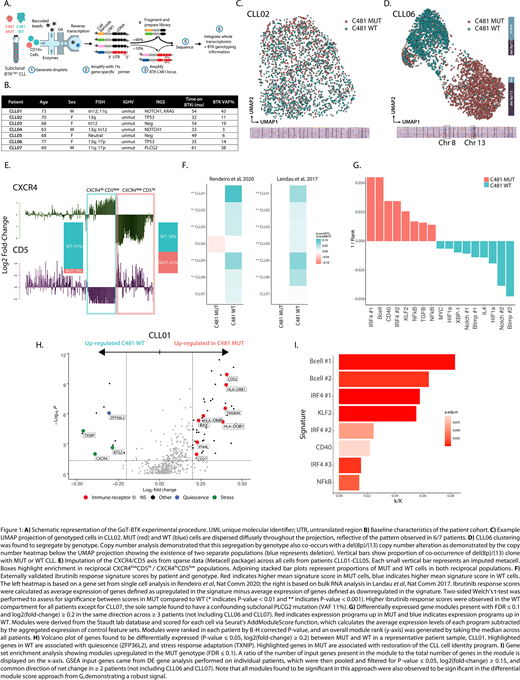
We applied GoT to 64,099 CD19+ cells across a cohort of seven patients with clinically progressive CLL found to have low-burden BTKC481 subclones (Fig. 1B). Samples were obtained at the time of progression and were screened for mutations in PLCG2. We genotyped 33.3% of cells, consistent with 34.0% of cells expressing BTK in whole transcriptome data. Clustering of the gene expression profiles showed that MUT and WT clones did not segregate by genotype in most (6/7) patients (Fig. 1C), implying a large degree of transcriptional similarity between MUT and WT cells, and further highlighting the need for multi-omics single-cell sequencing to directly link genotype to phenotype in this context. We note that the single exception with distinct genotypic clustering (CLL06, Fig. 1D) was driven by co-occurring large chromosomal aberrations within the MUT cells, including del(8p), which is associated with BTKi resistance [Burger et al, Nat Comm 2016].
CLL cells are known to cycle between the peripheral blood and protective microenvironmental niches, which is a process modulated by BTKi. We applied CXCR4/CD5 expression as a read out to these migration patterns [Chen et al, Leukemia 2016], and observed that MUT cells were comparatively enriched in CXCR4lowCD5hi CLL, which is associated with recent emigration from the stromal niche and increased BCR signaling (Fig. 1E). Conversely, WT cells were the majority of CXCR4hiCD5low CLL, which is associated with a resting peripheral cellular state and BCR downregulation, suggesting WT quiescence with limited stromal support.
To explore the phenotypic changes associated with BTK mutations, we first measured the activity of reported BTKi response expression signatures. WT cells showed higher mean response scores compared to MUT (Fig. 1F), demonstrating that the WT cells preserve BTKi responsiveness. To unbiasedly interrogate transcriptional differences, we applied a set of 336 predefined gene modules representing B-cell cellular functions and processes. Enriched modules in MUT cells implicated restoration of B-cell receptor (BCR) signaling and recovery of CLL cell identity (NF-kB, IRF4, CD40, Fig. 1G), a finding also supported by de novo differential gene expression analysis (Fig. 1H), and gene set enrichment of differentially expressed genes (Fig. 1I). In contrast, WT cells showed increases in modules related to quiescence, hypoxia, cellular stress (HIF-1α, XBP1) and terminal B-cell development (Blimp-1). Intriguingly, changes in gene targets associated with restoration of NOTCH1 and IL4 activity were also observed. As these pathways are impaired in patients responding to BTKi and implicated in resistance [Del Papa et al, CCR 2019; Chen et al, ASH 2019], their re-emergence may reflect a cytoprotective strategy.
In summary, we utilized multi-omics single-cell sequencing to identify the distinct transcriptional programs of admixed MUT and WT cells in subclonal BTKC481 progression. MUT cells showed robust escape from BTKi inhibition via increased immune receptor signaling and restoration of CLL cell identity program, while WT cells demonstrated a signature of hypoxia and stress response, with NOTCH1 and IL4 activation implicated as mechanisms of clonal persistence.
Trisal:Celgene: Current Employment, Current equity holder in publicly-traded company. Gandhi:Celgene: Current Employment, Current equity holder in publicly-traded company. Wiestner:Pharmacyclics LLC, an AbbVie Company, Acerta, Merck, Nurix, Verastem, and Genmab: Research Funding; NIH: Patents & Royalties: NIH. Allan:Acerta, Genentech, Abbvie, Sunesis, Ascentage, Pharmacyclics, Janssen, AstraZeneca, BeiGene: Consultancy; Celgene, Genentech, Janssen, TG Therapeutics: Research Funding; Abbvie, Janssen, AstraZeneca, Pharmacyclics: Honoraria. Furman:Genentech: Consultancy; Beigene: Consultancy; AstraZeneca: Consultancy, Research Funding; Acerta: Consultancy; Verastem: Consultancy; Pharmacyclics: Consultancy; TG Therapeutics: Consultancy, Research Funding; Incyte: Consultancy; Janssen: Consultancy, Speakers Bureau; Oncotarget: Consultancy; Loxo Oncology: Consultancy; Abbvie: Consultancy; Sunesis: Consultancy. Landau:Bristol Myers Squibb: Research Funding; Illumina: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal